Дифференциальная диагностика
Если у новорожденного отмечается персистирующая гипогликемия, необходимо установить причину данного состояния. Прежде всего, нужно провести тщательную оценку клинического состояния новорожденного. Необходимо помнить, что неонатальная гипогликемия может быть проявлением наследственных синдромов, которые далеко не всегда дают развернутую клиническую картину в неонатальный период. Это относится даже к хорошо известным заболеваниям. Например, синдром Беквита-Видемана. Масштабные исследования показывают [186], что этот синдром является очень гетерогенным как клинически, так и генетически. Встречается он с частотой 1 на 13700, но указывают, что вполне вероятно у части больных с не ярко выраженными клиническими проявлениями он вообще не диагностируется. У больных, кроме гипогликемии в неонатальный период, могут отмечаться макросомия, макроглоссия, спланхомегалия (включая печень, почки, надпочечники, поджелудочную железу), "эмбриональные опухоли" (опухоль Вильмса, нейробластома, рабдомиосаркома и др.), врожденные пороки развития (пупочная грыжа, почечные структурные аномалии, диастаз прямых мышц живота) и т. д.
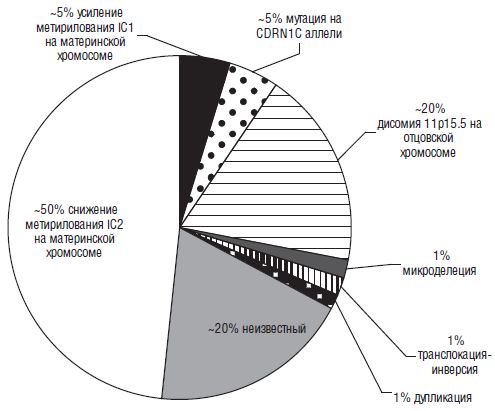
Рис. 9. Молекулярные нарушения при синдроме Беквита-Видемана (Shuman С. et al.5 2010 [186])
Но при рождении вся вышеперечисленная симптоматика встречается, повторимся еще раз, далеко не всегда. Например, макроглоссия и макросомия обычно присутствуют при рождении, но иногда начинают проявляться после рождения. Темпы роста начинают замедляться в возрасте около семи-восьми лет и т. д. Примерно 85 % больных с синдром Беквита-Видемана имеют спорадическую мутацию. 15 % детей имеют аутосомно-доминантный тип наследования, связанный с различными нарушениями на 11 хромосоме (11р 15.5). Наиболее часто выявляются следующие мутации (рис. 9). Интересно, что повышенный риск заболевания имеют дети, зачатые в результате методов вспомогательных репродуктивных технологий (ЭКО, ИКСИ).
И, на примере синдрома Беквита-Видемана, обратим внимание еще на один, возможно, один из важнейших аспектов неонатальной медицины: каждый ребенок, имеющий любое отклонение, даже самое незначительное, в неонатальный период как клинических, так и лабораторных показателей должен быть обследован в катамнезе. Он не должен исчезать из поля зрения врача, лучше того учреждения, где он родился или куда был переведен для лечения.
Детям с синдромом Беквита-Видемана рекомендуют [186] ежегодное ультразвуковое исследование почек до подросткового возраста для исключения нефрокальциноза, ультразвуковое исследования каждые три месяца органов брюшной полости для исключения опухолей, исследование концентрации альфа-фетопротепна каждые два-три месяца в течение первых четырех лет жизни с целью раннего выявления гепатобластомы и т. д. Представляется, что такие декретированные сроки и объем обследования должны быть определены для всех заболеваний, возникших в неонатальный период.
В последние десятилетия описано несколько форм семейного гиперинсулинизма, связанного с хромосомными и/или геномными мутациями [84, 94]. Сейчас этот термин заменил то, что раньше называлось "низидиобластоз", хотя и в настоящее время этот термин широко используется. Это название происходит от греческого "nesidion" - островок, "blastos" - зародышевые клетки, а также "osis" - опухоль. Этот термин подчеркивал, что при гистологическом исследовании имеется явное увеличение количества изолированных β-клеток и дезорганизация островков Лангерганса. Многочисленные исследования и сопоставление с контрольными группами показали, что указанные изменения не являются специфичными для данного заболевания, что привело к замене термина "низидиобластоз" на новый - "гиперинсулинизм".
Семейный гиперинсулинизм нередкое заболевание. В настоящее время оно зарегистрировано среди всех этнических групп. В Европе его частота составляет 1 случай на 15000 населения, а в центральной Финляндии и в Саудовской Аравии почти в 7 раз чаще (1:2500).
Заболевание характеризуется гипогликемией, которая может начинаться в неонатальный период, а иногда и позже, например в детском возрасте, с достаточно скудной симптоматикой, а, соответственно, и обусловленной этим сложностью диагностики. У новорожденных, как правило, заболевание манифестирует в течение первых двух суток жизни и протекает от выраженных форм с развернутой клинической симптоматикой гипогликемии до бессимптомных форм, когда лабораторные находки (снижение концентрации глюкозы крови) заставляют начать обследование ребенка. Даже в пределах одной семьи у ближайших родственников течение заболевания может варьировать от легкой до тяжелой степени. Иногда клиника связана с формой мутации.
Больные с аутосомно-рецессивной формой семейного гиперинсулинизма, обозначаемой в зарубежной литературе как FHI-KATФ, вызванной мутациями в АВСС8 или KCNJ11 , как правило, рождаются крупными к сроку гестации, развивают тяжелую гипогликемию в первые 48 часов жизни, резистентную к медикаментозной терапии и требуют для достижения нормогликемии хирургического лечения (резекции поджелудочной железы).
Больные с аутосомно-доминатной формой семейного гиперинсулинизма, в англоязычной литературе обозначаемой как FHI-GCK, вызванной мутациями в GCK , чаще всего манифестируют в возрасте одного года (диапазон от 2 дней до 30 лет). Как правило, данная форма, чаще всего протекает мягче, чем аутосомно-рецессивная, хотя некоторые больные нуждаются в агрессивной терапии.
Форма, сочетающая семейный гиперинсулинизм с гипераммониемией, чаще всего, протекает с умеренной гипераммониемией и поздним началом гипогликемии, как правило, не в периоде новорожденности [33, 140].
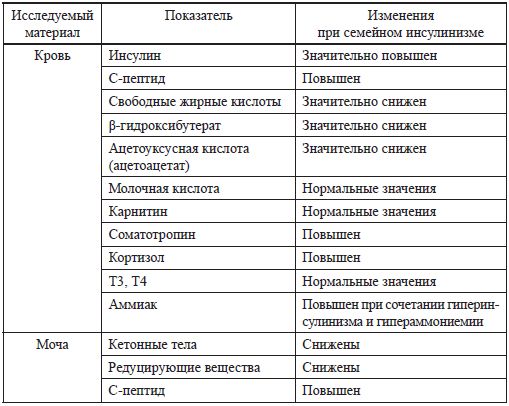
Таблица 9 Лабораторные тесты при семейном гиперинсулинизме (В. Glaser, 2010) [84]
Имеются и еще более редко встречающиеся формы гиперинсулинизма у новорожденных детей. У 40 % больных с семейным гиперинсулинизмом вообще, по крайней мере на сегодня, не удается идентифицировать мутации ни в одном из генов, связанных с развитием этого заболевания.
При всех формах гиперинсулинизма необходима консультация хирурга (после генетического и лучевого исследования) по поводу возможного оперативного лечения (необходимость тотальной или субтотальной резекции поджелудочной железы) и обследование на врожденные наследственные дефекты метаболизма (см. ниже).
В лабораторной диагностике, позволяющей подтвердить диагноз семейного гиперинсулинизма, имеют значение следующие тесты (табл. 9).
При гистологическом исследовании поджелудочной железы при семейном гиперинсулинизме выделяют два основных гистологических типа:
Диффузный - увеличение количества β-клеток по всей поджелудочной железе. Встречается примерно у 60–70 % больных. При этом сохранена архитектура поджелудочной железы. В-клетки имеют большие ядра, выраженную цитоплазму и гистологические признаки повышенной метаболической активности.
Фокусная аденоматозная гиперплазия поджелудочной железы. Встречается примерно у 30–40 % больных с семейным гиперинсулинизмом. Отмечаются очаговые изменения в тканях поджелудочной железы, а остальные ткани железы являются функционально и гистологически нормальными. В отличие от опухолей (истинных аденом) макроскопически фокальные изменения не видны. В-клетки, вне очагов поражения, имеют маленькое ядро и цитоплазму, достаточно редко в них имеются гистологические признаки, свидетельствующие о том, что они функционально подавлены и не могут вырабатывать инсулин. Сканирование с помощью позитронно-эмиссионной томографии успешно используется для предоперационной локализации очаговых поражений [165, 152].
В настоящее время возможна пренатальная и предимплантационная генетическая диагностика для выявления заболевания у эмбриона или плода, но для этого необходима предварительная идентификация мутаций у родителей.
Дифференциальную диагностику семейного гиперинсулинизма необходимо проводить с рядом заболеваний, сопровождающихся гипогликемией.
Около 30 % детей с синдромом Беквита-Видемана имеют гиперин-сулинизм, приводящий к развитию гипогликемии, но она обычно течет менее злокачественно, по-сравнению с синдромом семейного инсулинизма, и поддается медикаментозной терапии.
Врожденные дефициты кортизола или гормона роста могут иметь в клинической картине эпизоды гипогликемии, но она также обычно хорошо реагирует на заместительную гормональную терапию. Гипогликемия у новорожденных может проявляться как один признаков адреногенитального синдрома (дефицит 21-гидроксилазы). При этом необходимо помнить, что гипогликемия может быть одним из ранних симптомов при неклассических формах адреногенитального синдрома (дефицит 21-гидроксилазы с поздним началом).
Инсулиномы. Эти опухоли чрезвычайно редко встречаются у детей до года. Некоторые авторы [178] рассматривают семейные инсулиномы, как сложные эндокринные неоплазии 1-го типа, но они также редко встречаются у младенцев. Инсулиномы резко отличаются гистологически от низидиобластоза. Они являются истинными аденомами и состоят только из β-клеток.